
全国服务热线:13401021128
生物负荷 (Bioburden),也称为生菌数或负荷菌,通常指的是产品、原材料或表面上存在的活体微生物总数量 (如细菌、真菌、病毒等)。 它能有效评估和控制产品的微生物含量,不仅是确保产品品质的基石,更是保障患者与消费者安全的关键。
为什么生物负荷测试很重要?
生物负荷的来源是什么? 如何控制负荷菌 (bioburden control) ?
常见的生物负荷试验流程及方法?
「生物负荷试验」与“ 无菌试验”的差异?
国内外相关规范
生物负荷测试 (Bioburden testing),又称为生菌数测试 (total viable count, TVC) 或微生物限量(度)测试 (microbial limit testing),是制药与医疗产业中关键的品质控制过程 (quality control, QC),用于侦测及量化产品在灭菌前的活体微生物含量,能够反映原料品质、生产流程洁净度、污染控制状况,并作为制定灭菌参数及验证灭菌效果的依据。
全球主要法规机构,如美国食品药品监督管理局 (FDA) 和国际标准化组织 (ISO),都明确要求需执行生物负荷测试。 例如:
生物负荷测试被广泛应用于医疗器械、制药、生物制剂和食品与保健品等高风险产业,做为确保产品安全与合规的关键步骤。
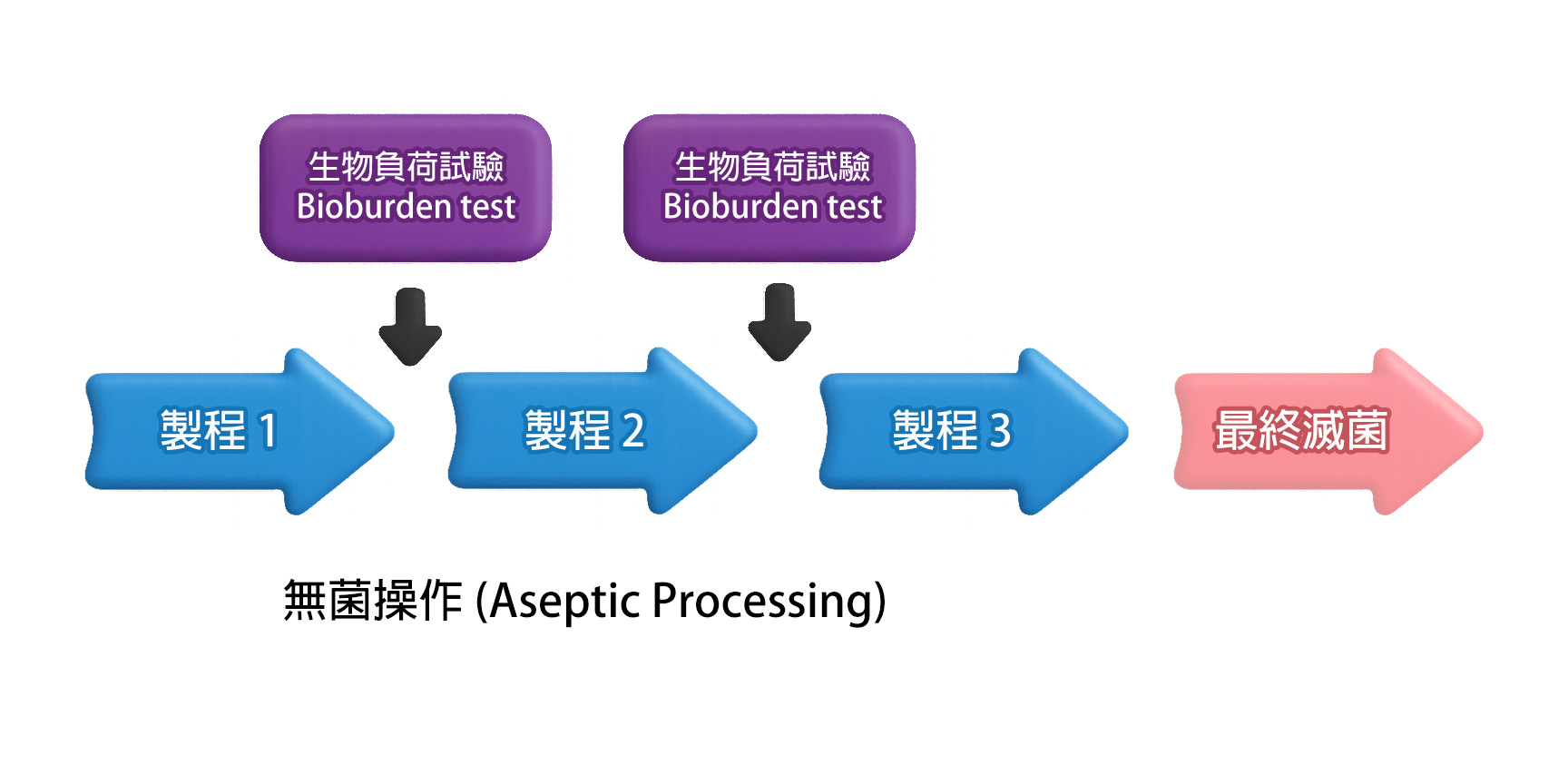
生物负荷测试一般在*终灭菌程序 (terminal sterilization) 前、或在无菌制程 (aseptic processing) 中「无菌过滤或充填步骤」前进行。 测试结果所反映的微生物含量,即为整体制程洁净度的指标。 因此,生物负荷测试被视为制程洁净验证与污染控制策略中的关键步骤,其代表意义如下:
微生物(microbial)可通过多种管道进入产品或原料中。 从原物料、人员、设备,乃至整个制造过程,都必须透过严格监控,使灭菌程序发挥*功效,制成使用安全无虞的产品。
常见的生物负荷来源及控制方式如下表所示:
| 来源 | 控制方式 |
| 原材料 | 对原材料进行微生物检测和控制 |
| 制造环境 | 维持洁净的生产环境,例如:使用无尘室、空气过滤系统等 |
| 人员 | 穿着无菌服装,并遵守严格的卫生程序 |
| 设备与工具 | 定期对设备和工作表面进行彻底清洁和消毒 |
| 制造过程 | 遵守无菌作规范,并选择产品合适的灭菌方法 |

1. 收集样品与处理:
依检品之物理特性选用适用的前处理方式,取得待测检液。 若无适用方法,则应建立适当之替代序程。
| 样品 | 检液配制 |
| 水溶性检品 | 以适当缓冲液或培养基溶解 |
| 非油性不溶于水检品 | 以适当缓冲液或培养基溶解,可添加界面活性剂 |
| 油性检品 | 检品溶解于适当酯类溶剂,再以预热稀释液稀释检品 |
| 喷雾式液体/固体检品 | 将容器中检品移入微孔滤膜过滤装置 |
| 贴片检品 | 具有黏性之面朝上放在无菌容器中,覆盖无菌多孔材料,再将贴布移入适当稀释液中,振盪至少30分钟 |
2. 选择适合的测试方法:
为了量化负荷菌,目前主流有四种定量分析方法,包含微孔滤膜过滤法 (Membrane Filtration Method)、倾注培养法 (又称混合稀释法,Pour Plate Method)、表面涂布法 (又称涂抹法,Spread Plate Method) 以及多重试管法 (又称*确数法, Most-Probable-Number, MPN),请根据检液特性选用适合的测试方法:
| 微孔滤膜过滤法(Membrane Filtration Method) | 培养皿法 / 平板法 (Plate Count Methods) | 多重试管法 (Most-Probable-Number, MPN) | ||
| 倾注平板法 (Pour Plate Method) | 表面涂布法 (Spread Plate Method) | |||
| 原理 | 检液以 0.45 μm 滤膜过滤后,将滤纸置于培养基上,进行培养 | 将检液与未凝固之培养基一起倒入培养皿中,进行培养 | 将检液涂抹于培养基中,进行培养 | 将检液进行至少3个连续10倍之序列稀释,进行培养 |
| 适合样品 | 可过滤之大量液态、生物负荷较低的样品 | 无法过滤之样品,如凝胶、黏胶物质等 | 无法过滤之少量样品 | 生物负荷极低的产品 |
| 适用热敏感物质 | 是 | 否 | 是 | 是 |
| 准确度 | 高 | 高 | 高 | 低 |
| 培养时间 | 约 5~7 天 | 约 3~7 天 | 约 3~7 天 | 约 3~7 天 |
| 结果单位 | 菌落形成单位 (CFU) | 菌落形成单位 (CFU) | 菌落形成单位 (CFU) | 依*确数表推估 |
* 一般认为多重试管法 (MPN) 是*不准确的方法,因此本法仅于无其他方法适用时才予选用。
3. 结果判定:
根据试验结果判定产品是否符合法规标准,或用以评估*终灭菌所需之水平。
其中,微孔滤膜过滤法不仅是*常见的生物负荷检测方法之一,也可以移除检体中的干扰物质。 若待测样品中含有抑制微生物的生长与繁殖的成分,如*剂(antimicrobial agent)或其他具抑菌活性(antibacterial activity)的成分,可能会导致检测结果出现伪阴性(即样品中实际存在微生物,但因生长受抑制而未被检出)。 因此,当检体特性适合以微孔滤膜过滤法作时,该方法常被视为进行生物负荷试验的*技术。 其方法是将检液通过0.45μm的滤膜,捕捉大于孔径的微生物,再将滤膜转置至适当培养基中培养,观察菌落生长情形。
为加速过滤流程,ROCKER 提供多款真空过滤系统(vacuum filtration system),皆兼容0.45 μm滤膜,并可搭配不锈钢、塑料材质漏斗,专为生物负荷测试设计。
| 洛科 MultiVac 301 – MB – A 多连真空过滤系统 | 洛科 SolarVac 1201 – MB – T 旋转多连真空过滤系统 |
|
|
|
|
|
「生物负荷试验」与「无菌试验」的差异?
生物负荷试验 (bioburden testing) 与无菌试验 (sterility testing) 是医疗器械和制药行业中,确保产品无菌性的两个不同但相关的测试,其目的、执行时机和方法有所不同。
| 生物负荷试验 (bioburden testing) | 无菌试验 (sterility testing) | |
| 目的 | 量化并监控制程中的活体微生物总量 | 确定*终产品的无菌性 |
| 执行时机 | 产品*终灭菌前的制程中设立检查点 | 产品经*终灭菌后 |
| 作环境 | 一般层流柜 (laminar flow) | 无菌检测隔离器 (Isolator) |
| 洁净度 Class 100 (A级) | ||
| 测试方法 | 微孔滤膜过滤法 倾注平板法 表面涂布法 多重试管法 | 微孔滤膜过滤法 直接接种法 |
| 测试对象 | 未经灭菌前的产品 非要求无菌之产品 | *终产品 要求无菌之产品 |
| 样器类型 | 药品( 固体及液体) | 药品(固体及液体) 医疗器材 |
| 测试结果 | CFU 数值 (微生物含量) | 是/否无菌 |
| 结果应用 | 评估灭菌剂量和参数; 微生物监测和趋势分析、制程风险评估 | 验证产品无菌性,*终放行产品的依据之一 |
| 标准依据 | ISO 11737-1 USP <61> USP <62> | ISO 11737-2 USP <71> |
| 中华药典 | (7007.1) 微生物计数法 (7007.2) 特定微生物检验法 | (7001) 无菌试验法 |
总体而言,生物负荷试验是预防性和工艺控制的工具,用以了解和管理产品在灭菌前的微生物含量信息; 而无菌试验则是验证性和质量保证的工具,目的为确认*终产品在经过灭菌处理后是否符合无菌要求
延伸阅读:无菌试验
生物负荷的试验与结果判读须依据国际标准进行,以确保结果的准确性。 国际间常见标准包含:
ISO 11737-1、美国药典 USP<61>、USP<62>、USP <1229.3>,以及药品优良制造规范 GMP (Good Manufacturing Practice) 等。 除了上述国际规范外,中国台湾亦有本国标准及指导文件可参考,如'中华药典'、行政院颁布之'无菌作作业指导手册'等,提供实务作与符合稽查要求的参考依据。
值得注意的是,微生物限度会依产品类型、使用途径与灭菌方式不同而有所变动。 产品若为非灭菌品,则必须证明其生物负荷处于可接受范围,避免对人体造成危害。

